Clayton Wandishin
Rational and computational design of an antibody fragment to improve affinity and cross-reactivity to influenza hemagglutinin and neuraminidase by the incorporation of non-canonical amino acids
Project Abstract
Influenza virus depends upon the function of two surface proteins, hemagglutinin (HA) and neuraminidase (NA), to effectively infect host cells. HA is responsible for binding and host cell entry of the virus, while NA is responsible for viral egress. Notably, these two proteins with distinct function bind to the same cell-surface molecule, sialic acid (SIA).
The goal of this project is to characterize the minimal sialic acid binding epitope of influenza neuraminidase, and hemagglutinin, by examining the electrochemical environment, the structural conformation, and the chemical properties of binding partners. These data will provide information to rationally design antibodies that can either 1) bind more tightly to the protein epitope, 2) bind more varied protein epitopes, or 3) both. Chemical space probing of the SIA binding pocket in HA and NA will be accomplished through Protein Data Bank (PDB) sorting, identifying the specific chemical interactions that must be satisfied for SIA to bind. Rational design will be approached using existing experimental structures of antibodies to SIA binding sites of HA and NA as well as co-crystal structures of bound ligands, while the computational design will be accomplished through the use of rosetta’s multi-state design protocols. Additionally, incorporation of non-canonical amino acids (ncAA) will be used to increase the chemical breadth of all designed antibodies. The anticipated outcomes of this project are identification of a cross-reactive antibody that binds to the sialic acid binding pockets of both influenza HA and NA through rational design, and production of computationally designed antibodies to both HA and NA using multi-state design protocols.
Progress Report
PDB sorting and comparison by EON (electrostatics) and ROCS (structural similarity) has been done, which found the most similar HA/NA pair to be H5 hemagglutinin and N9 neuraminidase (92% homology). SIA binding site comparison of the two is shown in Figure 1. Additionally, rational design looking at the active site and comparing it with previous high-throughput screening data revealed a propensity for para-substituted aromatics as strong complement to the binding site, which has been mimicked by the incorporation of para-amino-phenylalanine at the tip of an HA antibody (8M2). A model of this is shown in Figure 1. Moreover, this Fab has been expressed in E. coli as both a WT and para-amino-phenylalanine substituted variant. The results of this expression via SDS-PAGE are shown in Figure 2. Currently, we are awaiting LC/MS results to unambiguously confirm site-specific incorporation of the ncAA in the Fab HCDR3.
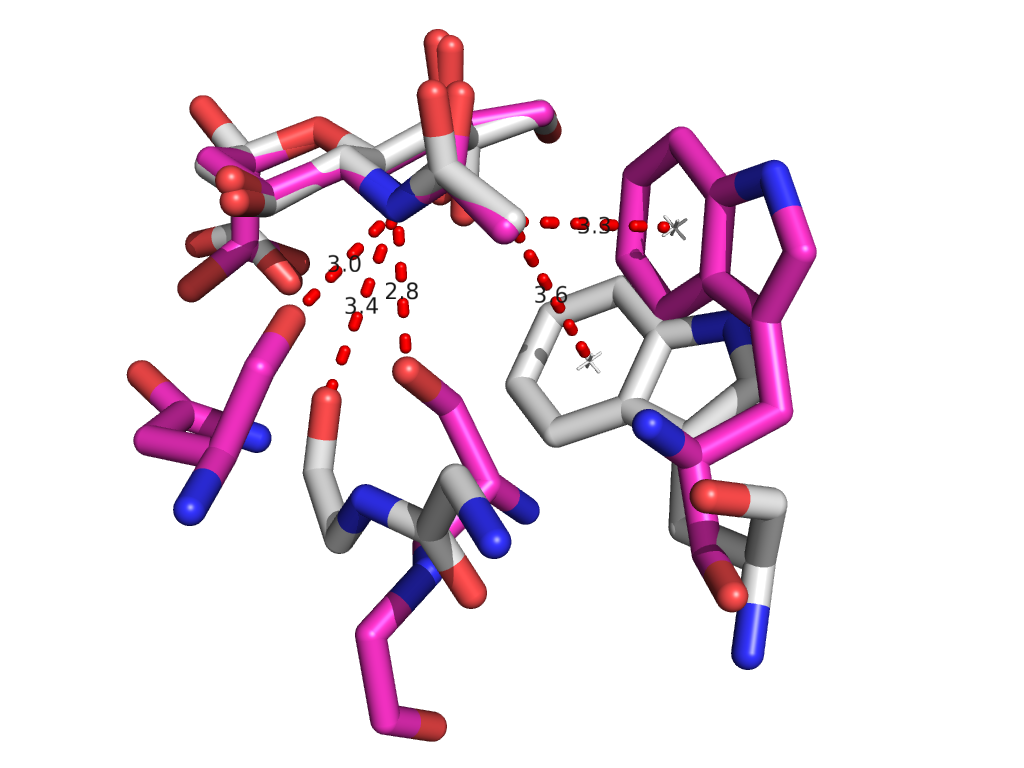
Figure 1a. PyMOL alignment showing the amino acid similarities and relative SIA conformation between an N9 neuraminidase (magenta) and an H5 hemagglutinin (gray)
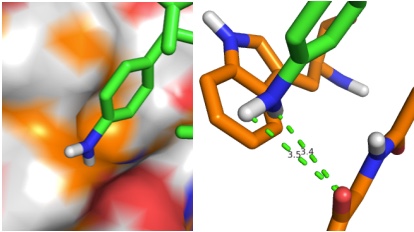
Figure 1b. Model of a para-amino-phenylalanine 8M2 variant at the minimal epitope of HA showing charge complementarity between the para-amino group and a backbone carbonyl of HA potentially creating a hydrogen bonding interaction
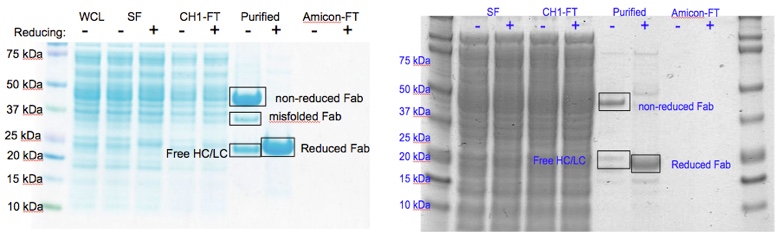
Figure 2. Figure 2. Comparison of SDS-PAGE gels for 8M2-WT Fab produced in E. coli (left) and 8M2-pAF Fab produced in E. coli (right) showing that for both WT and pAF the characteristic movement of the ~47 kDa band in the purified non-reducing lane to the ~22 kDa band in the purified reducing lane due to reduction of the Fab disulfide bonds. WCL=Whole Cell Lysate, SF=Soluble Fraction, CH1-FT=Anti-CH1 Column Flow-Thru, Purified=Anti-CH1 Column Elution post 30kDa Amicon concentration, Amicon-FT=30 kDa Amicon Spin Concentrator Flow-Thru, Fab=Antibody Fragment, HC=Fab Heavy Chain, LC=Fab Light Chain.
Primary: James E. Crowe, Jr.Secondary: Jens Meiler
Type of Trainee
Graduate student
©2024 Vanderbilt University ·
Site Development: University Web Communications