Gold-Benzenedithiolate-Gold Molecular Transport Junctions
In this article, we employed a hybrid molecular dynamics-Monte Carlo simulation scheme to study the impact of monolayer density, tip geometry, and temperature on the number, tilt angle, and bonding geometry of molecular junctions consisting of benzene-1,4-dithiolate (BDT) molecules chemically attached between gold nanotips. We found that monolayer density is an important determinant of the number of bridged molecules, with low monolayer density favoring the formation of multimolecule junctions and single-molecule junctions occurring most often at high monolayer density. We also showed that tip geometry and the presence of a monolayer, two factors often ignored in simulation, can have drastic effects on the structure of bridged molecules.
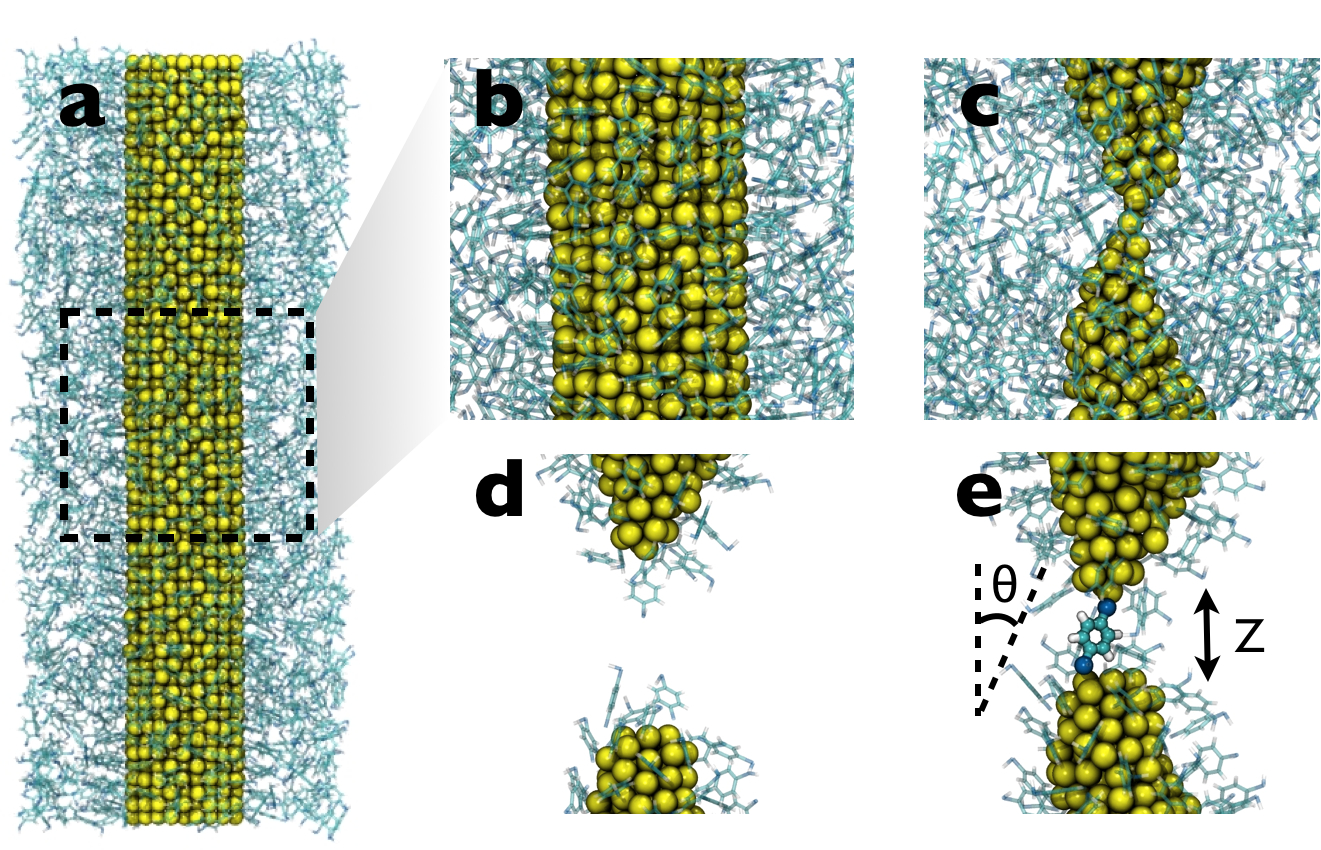
(a) BDT self-assembles onto an unstretched gold nanowire; a closeup is shown in (b). (c) Gold point contact in the necked region of the nanowire as a result of elongation. (d) Following nanowire rupture, the bulk BDT is evaporated from the simulation box. (e) The ruptured NW tips are brought together, resulting in the formation of a molecular junction.
In this letter, we investigated Au-BDT-Au junctions under elongation (i.e., break-junction simulations), calculating the conductance of the simulated junctions for direct comparison to experiment. In doing so we identified important structure-conductance relationships for these systems. Specifically, we showed that the formation of monatomic chains of Au atoms directly connected to BDT results in counterintuitive conductance increases during elongation. This result provides a detailed explanation for a poorly understood experimental mechanism, and may lead to improved control over mechanically responsive molecular devices.
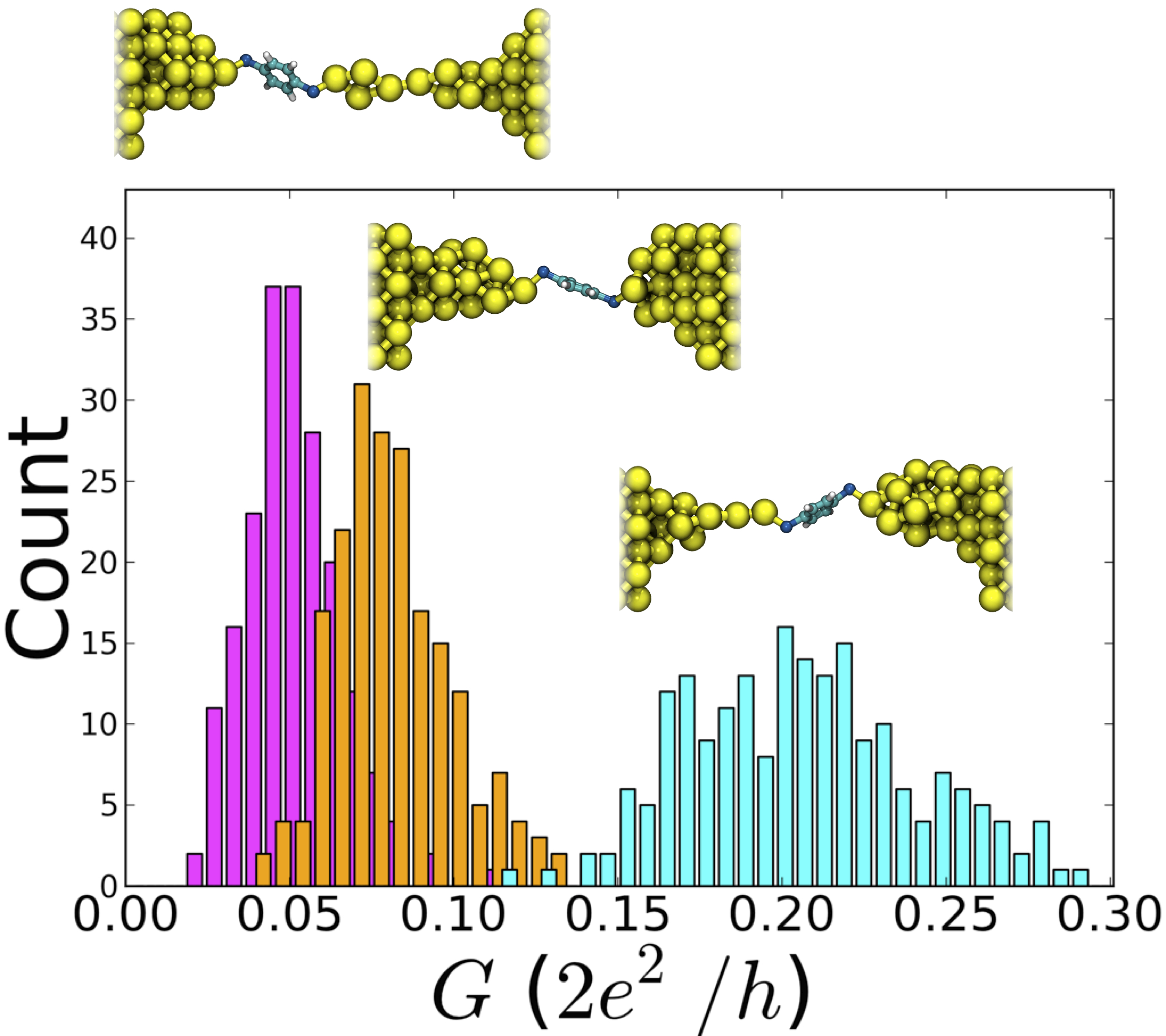
Conductance histograms of three thermally evolving Au-BDT-Au junctions. The right-most (light blue) histogram corresponds to a system with a monatomic chain, where negligible overlap with histograms for two other junctions is observed. The bin width is 0.006. Standard deviations of the histograms (from left to right) are 0.014, 0.018, and 0.034.
In a related letter, we investigated the structural origins of conductance fluctuations in Au-BDT-Au junctions. Conductance fluctuations are a major barrier to the construction of reliable molecular circuitry. Improving our understanding of the source of these fluctuations may enable the development of new strategies for limiting the magnitude of the fluctuations. We showed that structurally non-ideal junctions (i.e., those that have been stretched and deformed) exhibit larger conductance fluctuations than structurally ideal junctions. This result has important implications for break-junction experiments, where junction deformation may give rise to unacceptable structural changes that significantly alter the conductance behavior of the junction. Our findings also highlight the importance of incorporating realistic structural detail into theoretical calculations of molecular conductance.
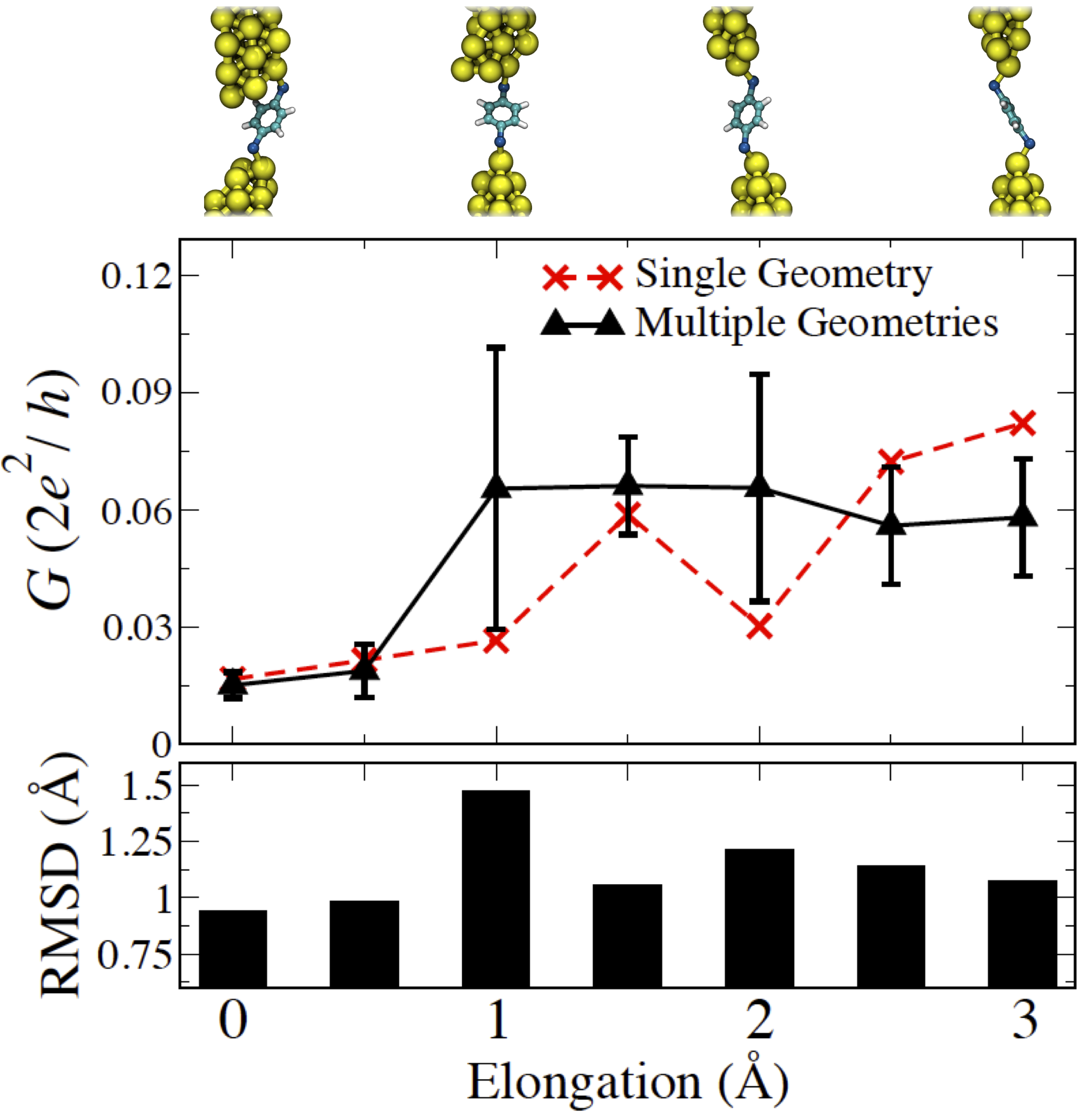
(Top) Thermally averaged and single-geometry conductance trace for Au-BDT-Au junction undergoing elongation. The initial junction geometries are shown above for every angstrom of elongation. (Bottom) Plot showing the root-mean-square deviation (the average value between the two tips) of the Au atom bonded to BDT.
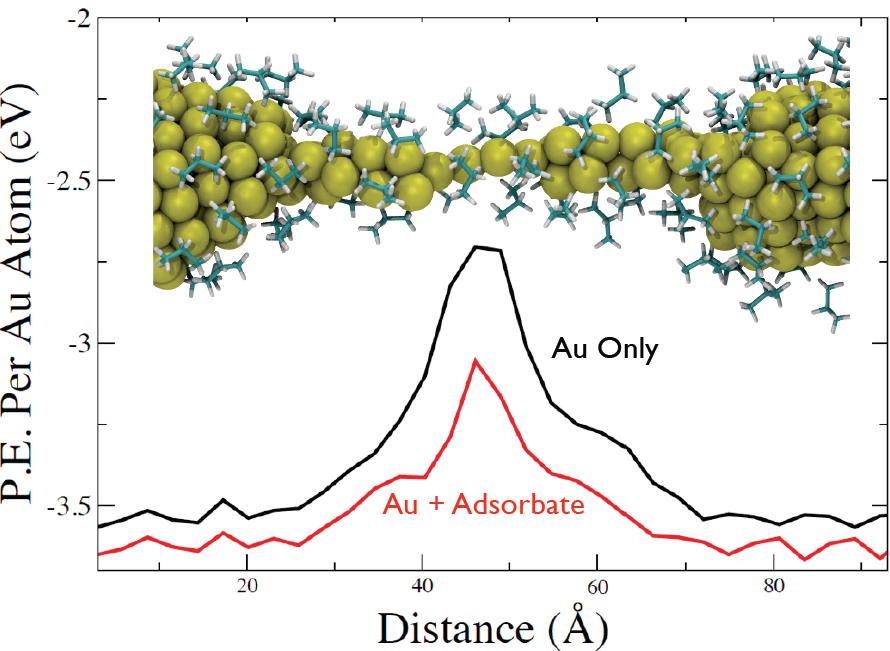
(Top Curve) The average potential energy acting on each Au atom in vacuum. (Bottom Curve) The average potential energy acting on each Au atom, including the contribution of both the Au-Au and adsorbate-Au interactions.
©2026 Vanderbilt University ·
Site Development: University Web Communications