Research
My current research focuses on the investigation of a host of nanoscale systems. Specifically, my work in the Cummings and McCabe groups focuses on (1) the simulation of nanowires and molecular junctions for nano- and molecular-electronics applications, (2) simulation and free energy calculations of nanoconfined fluids, (3) simulation of the phase behavior of grafted nanoparticles, (4) synthesis mimetic simulation of self-assembled monolayers, and (5) simulation and development of atomistic and coarse-grained models for the investigation of ceramide bilayer systems. My past graduate research focused on the simulation of the self-assembly of amphiphilic tethered nanoparticles into ordered crystalline and quasicrystalline structures, as well as the simulation and modeling micron-sized colloidal particles in direct collaboration with experiment. My current and past work includes a particular focus on the development and application of flexible order parameters based o
n shape matching.
Structure and Properties of Mechanically Deformed Nanowires and Molecular junctions:
Nano- and molecular-electronics systems, i.e., self-assembled electronic systems composed of atomic-scale contacts or single-molecules, hold significant promise for the creation of ultra dense, ultra fast electronic components. Understanding the correlations between structure and conductance is an important step towards the future design of future electronic devices.
In our work, we employ a combination of “classical” forcefields (both semi-emperical and reactive), shape matching order parameters, and density functional theory (DFT) conductance calculations to investigate the structure/property relationships in mechanical deformation of Au nanowires and the formation of molecular junctions composed of 1,4 benzene dithiol (BDT). Our approach to the problem is unique, in that we capture the entire process of junction formation, allowing the junctions to form naturally rather than starting from assumed, idealized starting configurations. Additionally, our approach enables us to capture thermal, environmental, and stochastic effects that would be present in experiment.
Related publications:
- French WR, Pervaje A, Santos A, Iacovella CR, Cummings PT, GPU-Enabled Large-Scale Statistical Study of Size-Dependent Failure in Elongating Gold Nanowires, submitted
- French WR, Iacovella CR, Rungger I, Souza A, Sanvito S, Cummings PT (2013) Atomistic Simulations of Highly Conductive Molecular Transport Junctions Under Realistic Conditions, Nanoscale, 5 pp 3654-3659
–DOI:10.1039/C3NR00459G (open access): arXiv:1303.5036 - French WR, Iacovella CR, Rungger I, Souza A, Sanvito S, Cummings PT (2013) Structural Origins of Conductance Fluctuations in Gold-Thiolate Molecular Transport Junctions , Journal of Physical Chemistry Letters, 4 pp 887–891
–DOI: 10.1021/jz4001104 - French WR, Iacovella CR, Cummings PT (2012) Large-Scale Atomistic Simulations of Environmental Effects on the Formation and Properties of Molecular Junctions, ACS Nano, 6, pp 2779–2789
– DOI:10.1021/nn300276m : arXiv:1202.3380 - Iacovella CR, French WR, Cook BG, Kent PRC, Cummings PT (2011) Role of Polytetrahedral Structures in the Elongation and Rupture of Gold Nanowires, ACS Nano, 5, pp 10065–10073
– DOI:10.1021/nn203941r : arXiv:1110.6396v1 - French WR, Iacovella CR, Cummings PT (2011) The Influence of Molecular Adsorption on Elongating Gold Nanowires, Journal of Physical Chemistry C, 115 pp 18422–18433
– DOI:10.1021/jp203837r : arXiv:1108.6251v1 - Iacovella CR, French WR, Cummings PT (2011) Flexible order parameters for quantifying the rate-dependent energy release mechanism of Au nanowires, Proceedings of SciDAC 2011, Denver, CO, July 10-14
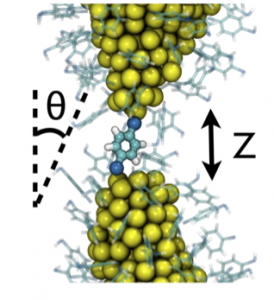 A ruptured Au NW, coated in BDT, with one BDT bridging the gap.
A ruptured Au NW, coated in BDT, with one BDT bridging the gap.
Nanoconfined Fluids:
Significant changes to the structure and properties of fluids can occur as they undergo nanoconfined. For 20+ years, there has been a disagreement in the literature regarding whether nanoconfined fluids undergo a first order phase transition to a crystal or a second-order constant vitrification as the pore size is reduced beyond a critical level. Within the group, we have focused on the application of simulation and theory to help understand the behavior of nanoconfined systems. In recent work, we adapted and applied the Einstein Crystal Method to calculate the absolute free energy of nanoconfined systems, to understand the transition from fluid to solid ordering.
Related publications:
- Wan L, Iacovella CR, Nguyen T, Docherty H, Cummings PT (2012) Confined fluid and the fluid-solid transition: The evidence from free energy calculations, Physical Review B, 86, pp 214105
– DOI:10.1103/PhysRevB.86.214105
- Cummings PT, Docherty H, Iacovella CR, Singh JK (2010) Phase transitions in nanoconfined fluids: The evidence from simulation and theory (Perspective), AICHE Journal, 56, pp 842–848
– DOI:10.1002/aic.12226
Self Assembly of Softmatter Crystals and Quasicrystals:
The ability to control the organization of soft matter into ordered arrangements is of particular interest in the nanoscale community. Dispersion vs. aggregation of nanoparticles and nanoscale aggregates can vastly alter the properties of a system. The creation of ordered arrays of the nanoscale can lead to other fundamental changes, such as the creation of photonic band gap materials or systems with catalytic activity. By examining grafted/tethered nanoparticles, we focused on the creation of generic “design rules” for the self-assembly of complex ordered structures, ranging from disordered micelles to ordered gyroid structures, lamella, and complex multicomponent crystalline materails
While crystalline structures are ubiquitous, and in many cases well understood, quasicrystals are much more rare and the mechanism for their formation in soft matter systems is not well understood. Quasicrystals are a unique class of ordered solids with long-range orientational ordering but no long-range periodicity (see Quasicrystals – Introduction by Ron Lifshitz). The discovery of quasicrystals in metallic alloys by Dan Schechtman and pioneering theoretical work by Dov Levine and Paul Steinhardt, forced scientists to re-evalutate the long standing “rules” of what it meant to be a solid. Dan Schechtman was recently awarded the Nobel Prize in chemistry for this discovery.

Time evolution of growing quasicrystal nucleus (yellow atoms) within a simulated supercooled liquid. Red atoms indicate icosahedral local ordering. (Credit: Aaron Keys, Chris Iacovella and Sharon Glotzer)
It has only been in recent years that the quasicrystals and related approximant structures have been observed in systems other than metallic alloys. In particular, softmatter micelle systems have recently been reported to form quasicrystalline structures. However, the formation of these quasicrystalline systems in softmatter has been sporadic and a clear theory or heuristic for designing them has been elusive. In recent work, performed by myself, Aaron Keys and Sharon Glotzer, we investigated the formation of soft matter quasicrystals that make use of simple design rules. In the manuscript Self Assembly of Soft Matter Quasicrystals and Their Approximants we proposed that quasicrystals can be formed based solely on controlling micelle functionalization and shape. This mechanism relies on promoting the formation of polytetrahedral structures via controling the conformational entropy of the outer layer (i.e., corona) of the micelle and suppressing crystallization through controlling shape (i.e., asphericity) of the micelles. An example dodecagonal quasicrystalline structure formed via simulation, using a simplified model of micelle, is shown below.
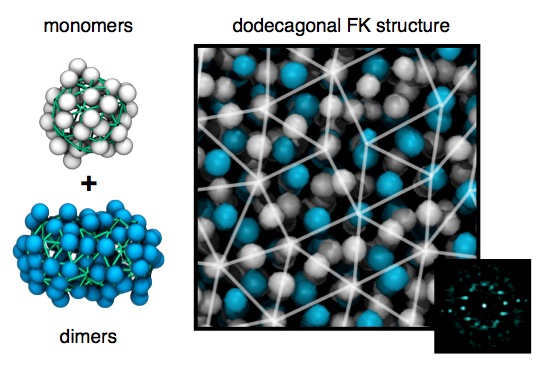
Dodecagonal Frank-Kasper (FK) quasicrystalline structure formed from a mixture of monomer and dimer micelles.
Related Publications:
- Iacovella CR*, Keys AS*, Glotzer SC (*authors contributed equally) (2011) Self Assembly of Soft Matter Quasicrystals and Their Approximants, Proceedings of the National Academy of Sciences, 108, pp 20935-20940
– DOI:10.1073/pnas.1019763108 : arXiv:1102.5589v2
- Phillips CL, Iacovella CR, Glotzer SC (2010) Stability of the double gyroid phase to nanoparticle polydispersity in polymer-tethered nanosphere systems, Soft Matter, 6, pp 1693–1703
– DOI:10.1039/b911140a
– Vir. J. Nan. Sci. & Tech. / Volume 21 / Issue 16 / SUPRAMOLECULAR AND BIOCHEMICAL ASSEMBLY
- Iacovella CR, Glotzer SC (2009) Phase Behavior of Ditethered Nanospheres, Soft Matter,5, pp 4492–4498
– DOI:10.1039/b909669h : arXiv:0907.5015v1
– Vir. J. Nan. Sci. & Tech. / Volume 20 / Issue 20 / SUPRAMOLECULAR AND BIOCHEMICAL ASSEMBLY
- Iacovella CR, Glotzer SC (2009) Complex crystal structures formed by the self assembly of di-tethered nanospheres, Nano Letters, 9, pp 1206–1211
– DOI:10.1021/nl900051u : arXiv:0901.0957v1
- Iacovella CR, Horsch MA, Glotzer SC (2008) Local ordering of polymer-tethered nanospheres and nanorods and the stabilization of the double gyroid phase, The Journal of Chemical Physics, 129, pp 044902
– DOI:10.1063/1.2953581 : arXiv:0806.0842v1
- Iacovella CR, Keys AS, Horsch MA, Glotzer SC, (2007) Icosahedral packing of polymer-tethered nanospheres and stabilization of the gyroid phase, Physical Review E (Rapid Communication), 75, pp 040801(R)
– DOI:10.1103/PhysRevE.75.040801 : arXiv:cond-mat/0702224v2
- Glotzer SC, Horsch MA, Iacovella CR, Zhang ZL, Chan ER, Zhang X, (2005) Self-assembly of anisotropic tethered nanoparticle shape amphiphiles, Current Opinion in Colloid & Interface Science, 10, pp 287–295
– DOI:10.1016/j.cocis.2005.09.011
- Iacovella CR, Horsch MA, Zhang ZL, Glotzer SC, (2005) Phase diagrams of self-assembled mono-tethered nanospheres from molecular simulation and comparison to surfactants, Langmuir, 21, pp 9498–9494
– DOI:10.1021/la051035l
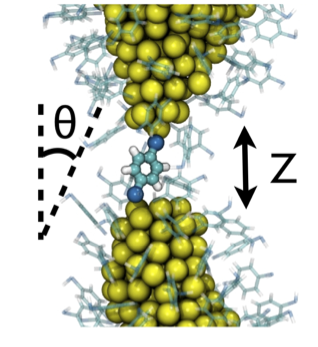 A ruptured Au NW, coated in BDT, with one BDT bridging the gap.
A ruptured Au NW, coated in BDT, with one BDT bridging the gap.
Nanoconfined Fluids:
Significant changes to the structure and properties of fluids can occur as they undergo nanoconfined. For 20+ years, there has been a disagreement in the literature regarding whether nanoconfined fluids undergo a first order phase transition to a crystal or a second-order constant vitrification as the pore size is reduced beyond a critical level. Within the group, we have focused on the application of simulation and theory to help understand the behavior of nanoconfined systems. In recent work, we adapted and applied the Einstein Crystal Method to calculate the absolute free energy of nanoconfined systems, to understand the transition from fluid to solid ordering.
Related publications:
- Wan L, Iacovella CR, Nguyen T, Docherty H, Cummings PT (2012) Confined fluid and the fluid-solid transition: The evidence from free energy calculations, Physical Review B, 86, pp 214105
– DOI:10.1103/PhysRevB.86.214105 - Cummings PT, Docherty H, Iacovella CR, Singh JK (2010) Phase transitions in nanoconfined fluids: The evidence from simulation and theory (Perspective), AICHE Journal, 56, pp 842–848
– DOI:10.1002/aic.12226
Self Assembly of Softmatter Crystals and Quasicrystals:
The ability to control the organization of soft matter into ordered arrangements is of particular interest in the nanoscale community. Dispersion vs. aggregation of nanoparticles and nanoscale aggregates can vastly alter the properties of a system. The creation of ordered arrays of the nanoscale can lead to other fundamental changes, such as the creation of photonic band gap materials or systems with catalytic activity. By examining grafted/tethered nanoparticles, we focused on the creation of generic “design rules” for the self-assembly of complex ordered structures, ranging from disordered micelles to ordered gyroid structures, lamella, and complex multicomponent crystalline materails
While crystalline structures are ubiquitous, and in many cases well understood, quasicrystals are much more rare and the mechanism for their formation in soft matter systems is not well understood. Quasicrystals are a unique class of ordered solids with long-range orientational ordering but no long-range periodicity (see Quasicrystals – Introduction by Ron Lifshitz). The discovery of quasicrystals in metallic alloys by Dan Schechtman and pioneering theoretical work by Dov Levine and Paul Steinhardt, forced scientists to re-evalutate the long standing “rules” of what it meant to be a solid. Dan Schechtman was recently awarded the Nobel Prize in chemistry for this discovery.
Time evolution of growing quasicrystal nucleus (yellow atoms) within a simulated supercooled liquid. Red atoms indicate icosahedral local ordering. (Credit: Aaron Keys, Chris Iacovella and Sharon Glotzer)
It has only been in recent years that the quasicrystals and related approximant structures have been observed in systems other than metallic alloys. In particular, softmatter micelle systems have recently been reported to form quasicrystalline structures. However, the formation of these quasicrystalline systems in softmatter has been sporadic and a clear theory or heuristic for designing them has been elusive. In recent work, performed by myself, Aaron Keys and Sharon Glotzer, we investigated the formation of soft matter quasicrystals that make use of simple design rules. In the manuscript Self Assembly of Soft Matter Quasicrystals and Their Approximants we proposed that quasicrystals can be formed based solely on controlling micelle functionalization and shape. This mechanism relies on promoting the formation of polytetrahedral structures via controling the conformational entropy of the outer layer (i.e., corona) of the micelle and suppressing crystallization through controlling shape (i.e., asphericity) of the micelles. An example dodecagonal quasicrystalline structure formed via simulation, using a simplified model of micelle, is shown below.
Dodecagonal Frank-Kasper (FK) quasicrystalline structure formed from a mixture of monomer and dimer micelles.
Related Publications:
- Iacovella CR*, Keys AS*, Glotzer SC (*authors contributed equally) (2011) Self Assembly of Soft Matter Quasicrystals and Their Approximants, Proceedings of the National Academy of Sciences, 108, pp 20935-20940
– DOI:10.1073/pnas.1019763108 : arXiv:1102.5589v2
- Phillips CL, Iacovella CR, Glotzer SC (2010) Stability of the double gyroid phase to nanoparticle polydispersity in polymer-tethered nanosphere systems, Soft Matter, 6, pp 1693–1703
– DOI:10.1039/b911140a
– Vir. J. Nan. Sci. & Tech. / Volume 21 / Issue 16 / SUPRAMOLECULAR AND BIOCHEMICAL ASSEMBLY - Iacovella CR, Glotzer SC (2009) Phase Behavior of Ditethered Nanospheres, Soft Matter,5, pp 4492–4498
– DOI:10.1039/b909669h : arXiv:0907.5015v1
– Vir. J. Nan. Sci. & Tech. / Volume 20 / Issue 20 / SUPRAMOLECULAR AND BIOCHEMICAL ASSEMBLY - Iacovella CR, Glotzer SC (2009) Complex crystal structures formed by the self assembly of di-tethered nanospheres, Nano Letters, 9, pp 1206–1211
– DOI:10.1021/nl900051u : arXiv:0901.0957v1 - Iacovella CR, Horsch MA, Glotzer SC (2008) Local ordering of polymer-tethered nanospheres and nanorods and the stabilization of the double gyroid phase, The Journal of Chemical Physics, 129, pp 044902
– DOI:10.1063/1.2953581 : arXiv:0806.0842v1
- Iacovella CR, Keys AS, Horsch MA, Glotzer SC, (2007) Icosahedral packing of polymer-tethered nanospheres and stabilization of the gyroid phase, Physical Review E (Rapid Communication), 75, pp 040801(R)
– DOI:10.1103/PhysRevE.75.040801 : arXiv:cond-mat/0702224v2 - Glotzer SC, Horsch MA, Iacovella CR, Zhang ZL, Chan ER, Zhang X, (2005) Self-assembly of anisotropic tethered nanoparticle shape amphiphiles, Current Opinion in Colloid & Interface Science, 10, pp 287–295
– DOI:10.1016/j.cocis.2005.09.011 - Iacovella CR, Horsch MA, Zhang ZL, Glotzer SC, (2005) Phase diagrams of self-assembled mono-tethered nanospheres from molecular simulation and comparison to surfactants, Langmuir, 21, pp 9498–9494
– DOI:10.1021/la051035l
Connect with Vanderbilt
©2026 Vanderbilt University ·
Site Development: University Web Communications