Marion Sauer
Project
“Optimizing dynamic viral epitope identification and design of structurally dynamic glycoproteins”
Abstract
For many viral pathogens, neutralizing antibodies recognize and bind almost exclusively to one or two envelope glycoproteins, which undergo concerted conformational rearrangements to facilitate the mechanical opening of the host cell membrane during fusion. Viral attachment and fusion occurs over the course of many minutes, making intermediate conformations viable epitopes for neutralization. Current structural biology techniques, however, have limited success elucidating intermediate structures of glycoproteins during fusion. The limited knowledge of viral glycoprotein conformation rearrangements precludes the development of vaccines that target intermediate conformations of a single peptide, let alone account for the structural consequences of antigenic drift. This project will use two strategies that independently allow for the rapid estimation of both fusion intermediate conformation rearrangements and sequence variation of large, highly flexible proteins. The first strategy sets out to represent conformational rearrangements as a series of directed backbone perturbations, which can be used to depict a series of intermediate conformations. The second strategy applies a recently improved protein design algorithm, REstrained CONvergence (RECON) MultiSpecificity Design (MSD) to estimate the sequence variation of a peptide backbone that can adopt multiple conformations. Together, both strategies can provide a high throughput toolset to assess the structural and functional consequences of viral sequence variation and predict conserved epitopes, which are accessible only during intermediate stages of attachment and fusion.
Project Update
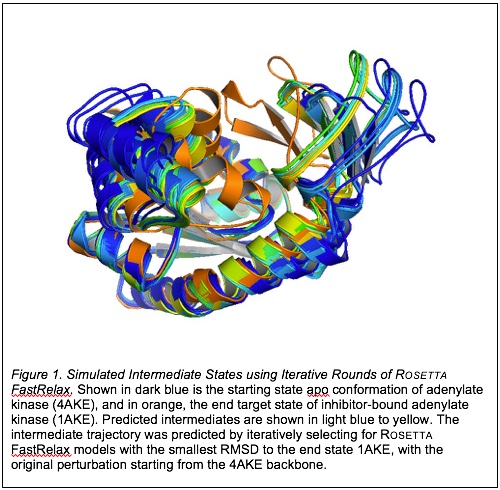
For the first strategy, rounds of Rosetta FastRelax were used to minimize the unbound adenylate kinase (AKE) structure (PDB ID: 4AKE), after each round selecting for either the lowest energy structure (in Rosetta Energy Units, or REU) or lowest RMSD to inhibitor-bound AKE (PDB ID: 1AKE) to guide perturbations that lead conformational rearrangements to a final state that resembles the end target 1AKE structure. Selecting for lowest RMSD to the end state 1AKE guided backbone perturbations toward the end state (Figure 1).
Based on the initial results, the quality metrics used to describe non-physical moves of large-scale conformational changes must be sensitive enough to select for small torsion angle deviations while also ensuring correct global conformation of each intermediate to avoid misrepresentation of an intermediate trajectory. Root mean square deviation (RMSD) is particularly insensitive to single atom superimposition fluctuations, whereas other quality metrics, such as dihedral angle RMS, global distance test, or contact strength difference, are better for discriminating against models with single aberrant distance fluctuations. Future directions of this project will validate which quality metrics should be implemented for iterative peptide backbone perturbation and selection sampling to estimate the lowest energy transition pathway, and to minimize the amount of sampling needed to converge on a single transition pathway.
For the second strategy, MPI-RECON MSD and Rosetta single-state design (SSD) was used to design the influenza HA2 monomer, Dengue E cleaved monomer, and the RSV F trimer using available conformations from the Protein Data Bank. A BLAST search was performed using the conformation with the least representation in the PDB, and each set of templates was chosen based on 100% sequence identity and zero gaps to the BLAST query. Each design run was repeated a hundred times, and the ten lowest total energy-scoring models were used to calculate the sequence tolerance.
Mentors
Primary: Jens Meiler and James Crowe
Secondary: Melanie Ohi and Ivelin Georgiev
Type of Trainee
Graduate Student
Publications
- Bender BJ, Cisneros A, Duran AM, et al. Protocols for Molecular Modeling with Rosetta3 and RosettaScripts. Biochemistry. 2016;55(34):4748-4763. doi:10.1021/acs.biochem.6b00444.
- Mousa JJ, Sauer MF, Sevy AM, et al. Structural basis for nonneutralizing antibody competition at antigenic site II of the respiratory syncytial virus fusion protein. Proc Natl Acad Sci U S A. 2016:1-10. doi:10.1073/pnas.1609449113.
©2024 Vanderbilt University ·
Site Development: University Web Communications