Nina Bozhanova

Project Abstract
Antibodies interact with antigens via a region on their surface made up of six complementarity-determining region (CDR) loops. The interactions between the surface patch on the antigenic molecule and the six CDR loops are mediated by reversible non-covalent interactions of the amino acids that involve shape complementarity, electrostatic interactions, hydrogen bonds, and hydrophobic interactions. The antibody structure and function are irrevocably linked to the sequence. We have access to millions of antibody sequences through next generation sequencing technology, however, it is not feasible to structurally model all of them to predict their interaction with antigens. Homology-based search is also limited. The goal of this project is to provide an effective approach that combines the accuracy of structure-based predictions with the speed of sequence-based methods for rapid selection of perspective novel antibody sequences that are structurally and functionally homologous to known antibodies for further detailed analysis. We propose to computationally evaluate a small random subset of sequences for their ability to mimic a given antibody in complex with an antigen using Rosetta, build position specific scoring matrix (PSSM), and then use it to rapidly filter and prioritize sequences for complete structural evaluation.
Project Update
We investigated different sized training datasets, the number of generated Rosetta models per sequence, and different strategies for PSSM scores calculation to find the best balance between accuracy and time used. Using 1200 sequences as a training set, ridge regression, and the 5 best models with the lowest total energy scores out of 10 generated models we managed to decrease the noise in residue scores between different training datasets and improve the convergence of the results.
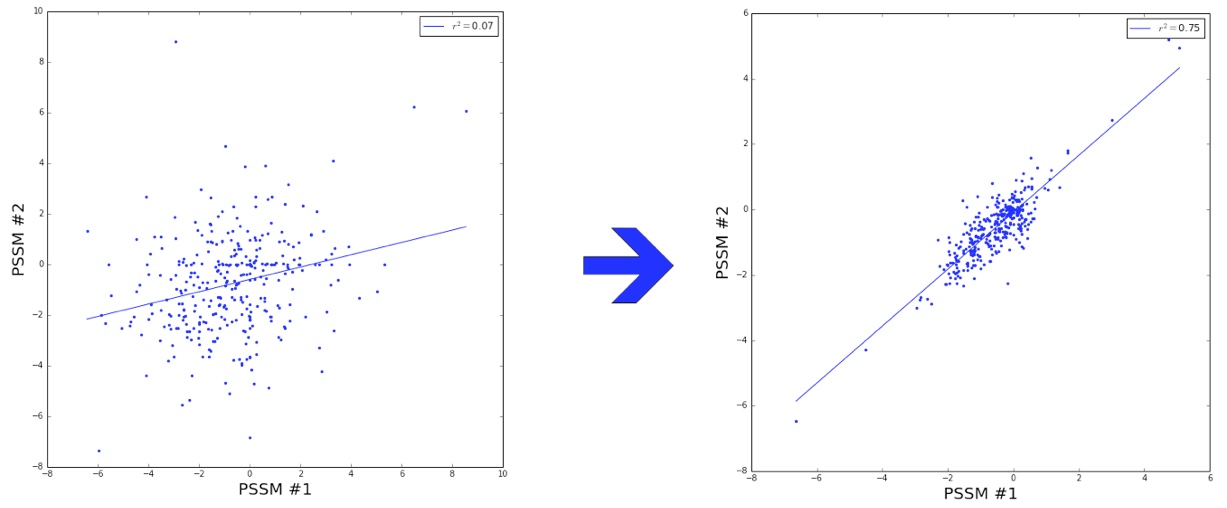
Notable accolades
Nina is 1 of 6 speakers at our annual Symposium on Modeling Immunity May 1, 2019. She will present “Functional antibodies in naive donors: Predicting binding through structural similarity”
Mentors
Primary: Jens Meiler
Secondary: James E. Crowe, Jr.
©2024 Vanderbilt University ·
Site Development: University Web Communications